care
Stars: 87
Automated creation and manipulation of chemical reaction networks (CRNs) in heterogeneous catalysis, powered by state-of-the-art ML evaluators and Julia-solvers for microkinetic modelling.
A collection of some of my projects, publications and designs.
Stars: 87
Automated creation and manipulation of chemical reaction networks (CRNs) in heterogeneous catalysis, powered by state-of-the-art ML evaluators and Julia-solvers for microkinetic modelling.
Stars: 7
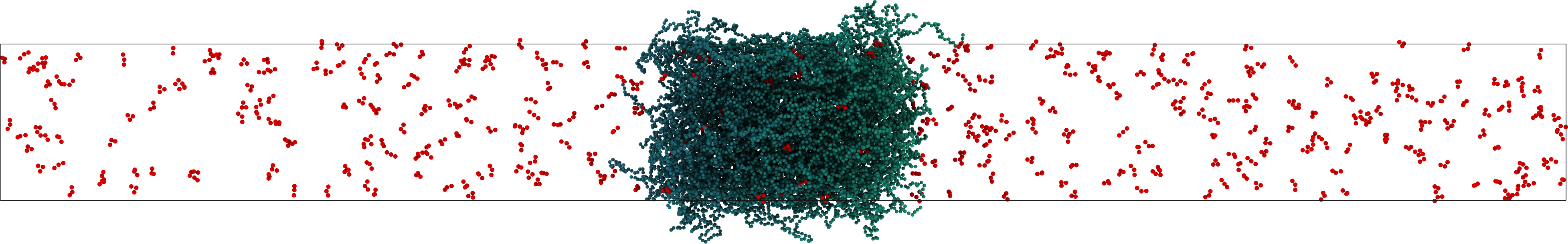
Tools to study LLPS in IDP systems using the OpenMM python API
Stars: 6
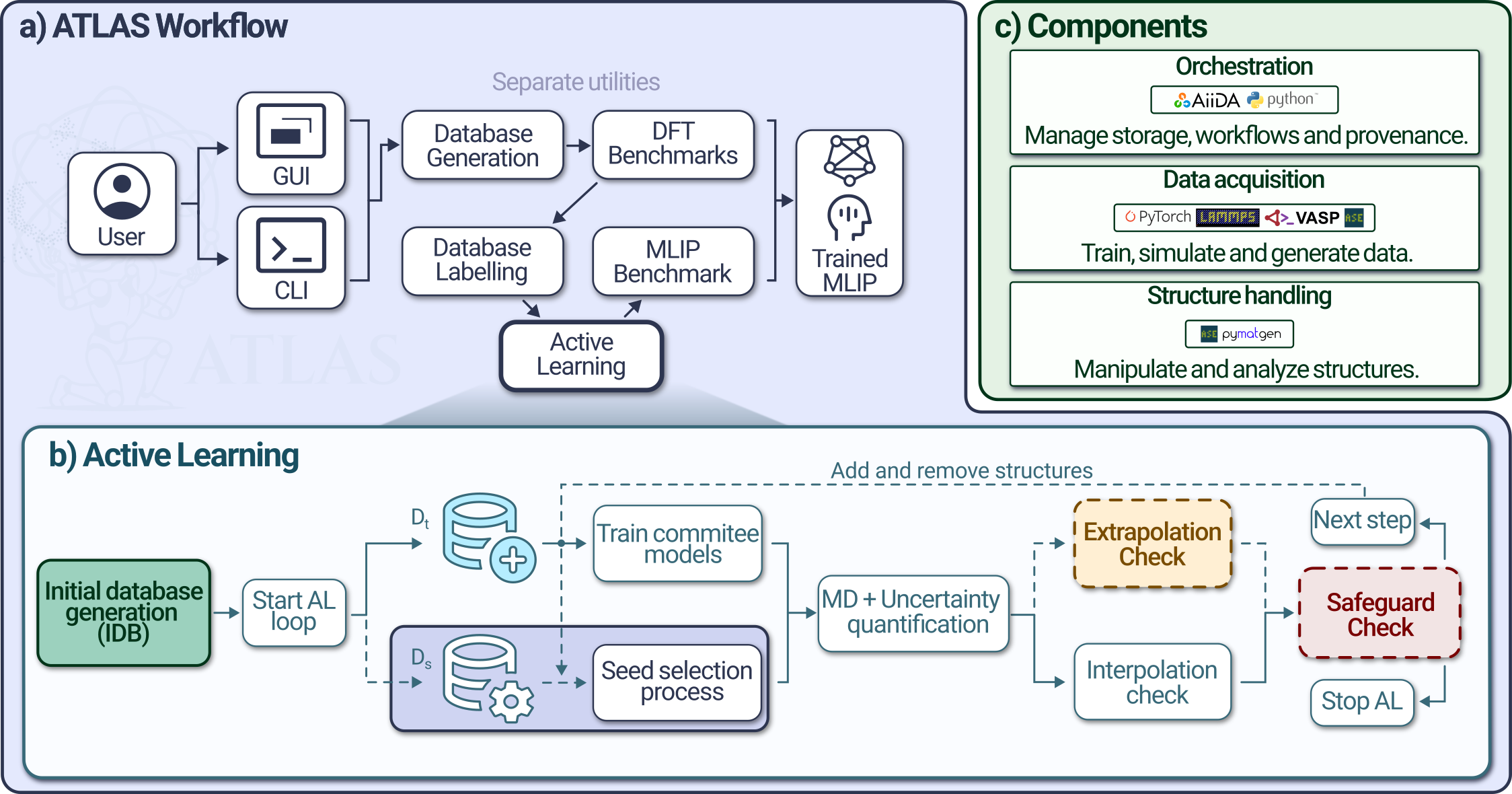
An automatic workflow for streamlining training and data generation for MLIPs via active learning, using latent-space aware design.
Year: 2024 | Citations: 410
The oxygen evolution reaction is the bottleneck to energy-efficient water-based electrolysis for the production of hydrogen and other solar fuels. In proton exchange membrane water electrolysis (PEMWE), precious metals have generally been necessary for the stable catalysis of this reaction. In this work, we report that delamination of cobalt tungstate enables high activity and durability through the stabilization of oxide and water-hydroxide networks of the lattice defects in acid. The resulting catalysts achieve lower overpotentials, a current density of 1.8 amperes per square centimeter at 2 volts, and stable operation up to 1 ampere per square centimeter in a PEMWE system at industrial conditions (80°C) at 1.77 volts; a threefold improvement in activity; and stable operation at 1 ampere per square centimeter over the course of 600 hours.
Year: 2025 | Citations: 12
Assessing the sustainability of plastic chemical recycling requires realistic feedstocks and catalysts designed within sustainability-led frameworks (Plastic-to-X). We link catalyst design and systems analysis to study hydrogenolysis of high-density polyethylene (virgin and bottle caps; Mw=100–200 kDa). We report Ru–Ni alloy nanoparticles (3–4 nm) supported on titania that yield up to 55% liquid C6–C45 products under optimized conditions, whereas monometallic Ru produces virtually no liquids Operando spectroscopy and simulations reveal structure sensitivity: backbone scission follows dehydrogenation and hydrogenation cycles at defective alloy sites formed in situ. Integrating these mechanistic insights with life cycle and techno-economic analyses indicates profitable processing of plastic caps over the optimal catalyst (2.5 wt% Ru, 5 wt% Ni) with substantially lower CO2 emissions even when using green H2. Furthermore, within the Plastic-to-X framework, we identify a minimum average chain length threshold of C11 for product distributions as a critical design metric to reconcile environmental and economic objectives.
Year: 2024 | Citations: 8
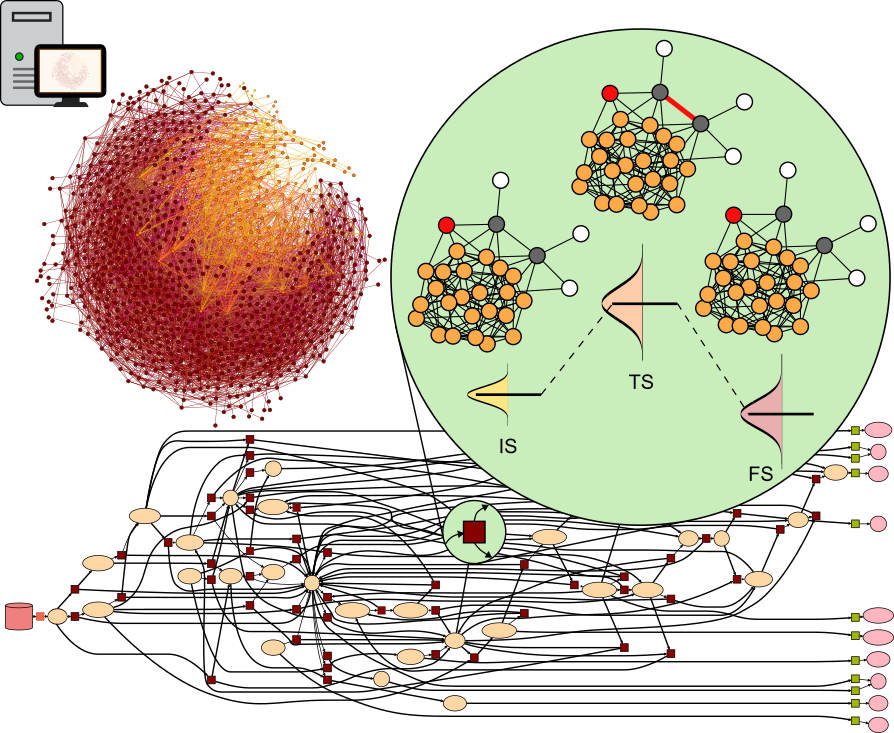
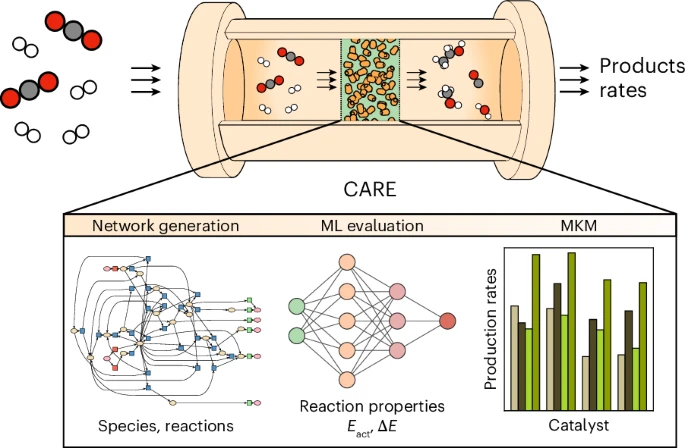
Process optimization in heterogeneous catalysis relies on the control of competing reactions. The reaction mechanisms based on chemical knowledge can be evaluated via density functional theory unveiling experimental catalytic trends. However, this approach finds its limits when applied to complex reaction networks or large molecules, disregarding alternative paths and rare events. Here we present CARE, a foundational model for catalysis on metal surfaces with a rule-based reaction network generator for CxHyOz species built with GAME-Net-UQ, a graph neural network with uncertainty quantification targeting thermodynamic and kinetic parameters, coupled to microkinetic modeling. CARE reproduces experimental activity trends in methanol decomposition, selectivity to C3 products in electrochemical reduction processes, and models the Fischer-Tropsch synthesis to C6 products, including 370k reactions, breaking the current limits of network exploration. This comprehensive model opens the path towards the exploration of thermal and electrocatalytic surface processes previously not amenable to atomistic simulations.
Year: 2026 | Citations: 5
The rationalization of catalytic processes relies on the fundamental understanding of competing reaction mechanisms driving reactants to products. The list of elementary steps composing the reaction networks is proposed based on chemical intuition and evaluated via density functional theory. This approach is limited by the size of the network and disregards alternative paths. Here we present the Catalytic Automated Reaction Evaluator (CARE), a flexible end-to-end framework for heterogeneous catalysis composed of (1) a rule-based reaction network generator, (2) a thermodynamic and kinetic parameter evaluator powered by state-of-the-art machine learning models and (3) a fast microkinetic solver. CARE reproduces the experimental activity trends in methanol decomposition, identifies the selectivity to C3 products in CO2 electroreduction and generates the Fischer–Tropsch synthesis mechanism including 370,000 reactions reaching C6 products. This comprehensive framework enables the exploration of thermal and electrocatalytic reactions previously not amenable to atomistic simulations.
Year: 2025 | Citations: 1
The design of low-temperature fuel cells and electrolyzers involving proton exchange membranes (PEMs) has been dominated by the exceptional performance and industrial dominance of Nafion exttrademark. With emerging regulations limiting the use of fluorinated compounds, the search for F-free alternatives must be accelerated. Here, we optimize a fluorine-free liquid crystalline poly(epichlorohydrin) membrane, based on the identification of key descriptors for proton mobility obtained using machine learning potentials derived from first-principles calculations for extensive molecular dynamics. Proton transfer is controlled by the interplay between water confinement and internal pore chemistry. This allows us to modify the active functional groups so that they directly participate in the proton diffusion process, increasing three times the diffusion rate in the membrane thus being close to Nafion standards. This work provides key descriptors for a targeted design approach essential for catalyzing the commercial development of next-generation fluorine-free PEMs.
Year: 2026 | Citations: 0
Machine learning interatomic potentials enable atomistic simulations at near first-principles accuracy. As model architectures mature, their reliability is increasingly constrained by training data quality. Here, we introduce Automated Training with Latent-space-Aware Sampling, ATLAS, a unified and multifunctional framework based on reversibly compressed latent space and online reliability verification. By co-designing diversity and efficiency criteria with a diversity-aware database generator and a manifold-aware active learning workflow, we steer exploitation toward sparse configurational regions in the structural space while preventing premature convergence. ATLAS produces compact datasets that allow MLIP training with high accuracy reducing up to 300 times fewer structures. To test ATLAS-trained MLIPs, we looked for high-temperature properties such as melting (physical) and decomposition (involving chemical reactions), demonstrating that our methodology consistently brings down prediction errors for temperature below 175 K, for Cu, CuZn and IrO2. ATLAS provides a principled and transferable strategy for building robust, similarly accurate and well-balanced MLIPs across diverse material classes.
Year: 2026 | Citations: 0
Metal–oxide interactions are ubiquitous in many technological applications and involve a complex interplay between the oxide support and the metal nanoparticle. Particularly, it has been proposed that in strong metal–support interaction, the defect chemistry affects the metal cluster morphology. Here we develop a physics-guided machine learning framework to decode these interactions using Pt7 and Pt13 representative of planar and tridimensional clusters, analyzing the impact of across oxygen vacancy concentrations of CeO2–x = 0–12.5% (528 configurations). Our models (R2 > 0.97) reveal that polaron swarms, rather than defect concentrations, predominantly control cluster shape and charge through size-dependent pathways. The framework yields quantitative design principles for defect-driven catalyst optimization and provides a general methodology for systematic mechanisms of metal–support interactions across diverse catalyst systems.

Year: 2020 | Citations: 0
The chemistry of enamines has gained importance in recent years due to the increased popularity of asymmetric organocatalysis using chiral secondary amines. The main goal of this dissertation is to gain insight, from a theoretical point of view, of the position of the equilibrium involved in the catalytic cycle of nitro-Michael reactions between the product enamine, containing a nitro group, and the starting aldehyde enamine.To do so, the energies of the species involved in the equilibria shown below were calculated, at different levels of theory. The calculations were performed with the Gaussian computational chemistry package and some scripts in Bash command language and in Python programming language were written to assist in the automatization of the process











